学校心臓検診は昭和48年の学校保健法施行規則の一部改正によって、定期健康診断の項目のひとつとしてその実施が義務づけられました。しかし、その実施方法については、定められたやり方はなく、各自治体に委ねられており、その対応は様々です。民間業者に丸投げして、心電図所見も自動判定のものがただ送られてくるだけで、その後のフォローもされていない自治体がたくさんあるのが現状ではないでしょうか。たつの市では、以前は循環器専門のY先生におんぶに抱っこ状態でほとんど頼りきっておりましたが、平成17年のたつの市合併に伴い、児童、生徒数が大幅に増えたことで、従来のシステムではなかなか対応が難しくなったため、医師会内に学校心臓検診委員会を立ち上げたわけです。
しかし、実際に新たに学校心臓検診のシステムを一から作ることは、本当に大変でした。たまたま、隣の姫路市医師会に立派な学校心臓検診委員会のモデルがあり、その委員の先生の助言が得られたので、なんとかそれっぽい感じで、たつの市でも学校心臓検診委員会がヨチヨチ歩きしだしたところです。

学校心臓検診の目的は、施行当初は心臓病の早期発見、治療することで、心疾患児に適切な指導を行い、出来るだけ健康な生活をできるようにし、突然死を予防することでした。しかし、最近では、ほとんどの先天性心疾患は入学前に診断・治療されています。小児循環器学の進歩は目覚ましく、救命も難しかった重症先天性心疾患の治療成績も以前とは比べ物にならず、学校生活を普通に楽しんでいることも少なくありません。しかし、一方で一見、健常児と変わらない先天性心疾患術後の児童・生徒が突然死したりすることもあり、現在では心臓手術後例や不整脈、川崎病、QT延長症候群、ブルガダ症候群、心筋疾患の発見とその生活管理指導が主眼となってきているが、同時に学童期に健やかな心身の発育、発達を遂げるためには身体運動は必要欠くべからざるものであり、過度の運動制限を課することには慎重でなければなりません。
学校心臓検診において、最も重要なことは精度管理です。学校心臓検診が精度高く実施されていれば二次検診が必要として抽出されるのは、全児童生徒の2〜5%とされています。先天性心疾患は出生1000人に対して8〜10人発生するとされています。後天性の心疾患では川崎病、不整脈ではWPW症候群が多く見られます。また、心肺蘇生法やAED(自動体外式除細動器)の普及で多くの人が救命される時代になっており、学校現場で児童生徒が倒れたときに、直ちに適切な処置が実施できる体制を普段から作っておくことも大切である。
不整脈
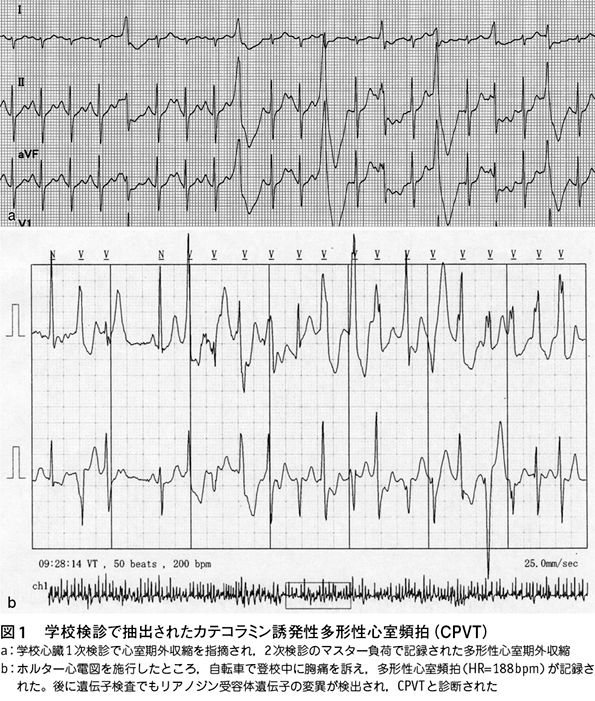
学校心臓検診(1次検診)で発見される不整脈で頻度が高いのは、期外収縮と洞性徐脈、房室ブロックです。2次検診以降では12誘導心電図、運動負荷心電図、ホルター心電図、心エコー等を行い、運動制限が必要なハイリスク例については専門医療機関へ紹介します。見逃してはいけない所見は、運動誘発性、QRS波の多形性、上室・心室頻拍、失神などの症状,突然死や致死的不整脈の家族歴です。先天性QT延長症候群(LQTS)は心電図で診断できる重要な遺伝性不整脈であり、適切な診断と生活管理、必要に応じたβ遮断薬投与により致死的イベントを予防できることができます。また、安静時心電図が正常であっても、運動時,精神的緊張時に失神する児童学童の中に、カテコラミン誘発性多形性心室頻拍(CPVT)という重症な遺伝性不整脈があります。
心室性期外収縮
単形性で出現数が少なく,運動負荷で消失するか増加しないものは、運動制限不要で1~2年毎に経過観察し、増悪傾向がなければ管理不要でもかまいません。単形性非持続性心室頻拍(心室期外収縮3連発以上100連発未満,または持続時間30秒以内)単形性持続性心室頻拍(100連発以上または持続時間30秒以上)多形性心室頻拍(2種類以上のQRS波形を示す心室頻拍)はいずれも、特に心拍数>150/分の場合、運動誘発性の場合、失神、眼前暗黒感、動悸等の症状を伴う場合は、専門医療機関へ紹介します。
カテコラミン誘発性多形性心室頻拍(CPVT)
予後不良の遺伝性不整脈で、運動や精神的緊張に際して多形性または二方向性心室頻拍や心室細動をきたし、失神や突然死を呈します。平均発症年齢は10歳で、安静時心電図は、徐脈傾向以外に異常を示さないことも多いため、失神があっても立ちくらみや起立性低血圧、てんかんなどと診断されていることがあります。失神、眼前暗黒感、動悸や強い胸痛などの症状(特に失神は致死的不整脈の重要な徴候として認識すべき)や突然死の家族歴についての注意深い問診が重要です。たとえば心室期外収縮ではQRS波形が多形性を示す場合、運動で増加する場合、心室頻拍を伴う場合などに注意が必要です。運動負荷で初めて診断がつくこともあり、心拍数≧150/分を目安に十分な負荷をかけることも重要です。

促進型心室固有調律(accelerated idio-ventricular rhythm:AIVR)
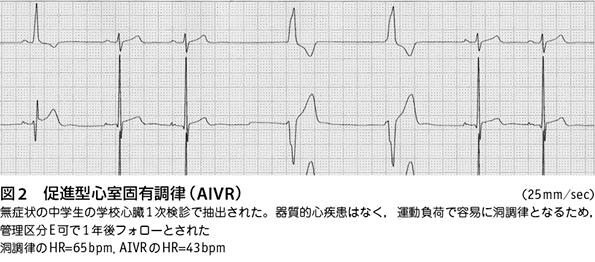 心室固有の拍数(30~40/分)よりも速く,洞調律前後の心拍数を示すことが多いため,促進型と言われる。多くは無症状で,検診で偶然に発見される。経験の少ない検診担当者は判定に迷うことがあるが,器質的心疾患を伴わず運動負荷で容易に洞調律に復帰するものは予後良好で,経過観察のみとなることが多い。心拍数>100/分のときは専門医療機関へ紹介
心室固有の拍数(30~40/分)よりも速く,洞調律前後の心拍数を示すことが多いため,促進型と言われる。多くは無症状で,検診で偶然に発見される。経験の少ない検診担当者は判定に迷うことがあるが,器質的心疾患を伴わず運動負荷で容易に洞調律に復帰するものは予後良好で,経過観察のみとなることが多い。心拍数>100/分のときは専門医療機関へ紹介
し,心室期外収縮・頻拍が混じるものはその管理に準じる。
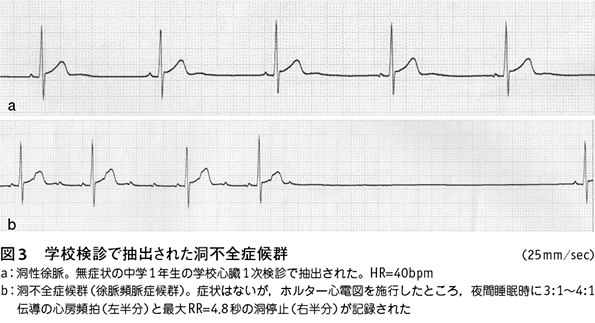
洞不全症候群
房室ブロック
小学生ではPR時間>0.24秒,中高生ではPR時間>0.28秒を2次検診以降への抽出基準とする。
(2)Wenckebach型2度房室ブロック
PR時間が徐々に延長し心室への伝導が途切れるタイプで、比較的多くみられる。運動負荷心電図,ホルター心電図を施行し,活動時に正常房室伝導になるものは管理不要でもよい。運動負荷で2度から回復しない場合またはブロックの程度が進行する場合は運動制限が必要である。
(3)MobitzⅡ型2度房室ブロック、2:1房室ブロック、高度房室ブロック (P波3つのうち2つ以上が心室に伝導しない)、完全房室ブロック
運動制限が必要なことも多く、専門医療機関へ紹介し電気生理検査やペースメーカーを含めた管理指針を検討する。特に失神、易疲労感などの症状がある場合は早期に対応する必要がある。
WPW症候群
顕性型は心電図の特徴的波形(PR時間短縮,デルタ波,QRS幅の拡大)から容易に診断されるが、間欠性WPW症候群(Wolff-Parkinson-White syndrome)では心室期外収縮などと間違われていることがある。無症状であると心電図検診で初めて診断されるため、WPW症候群を持っている人の中に、心臓の中に構造の異常、例えば孔があいているとか、ちょっと弁のつき方がおかしい、逆流があるという疾患もありますので、初回は心エコーにより器質的基礎心疾患(主にEbstein病と肥大型心筋症)をスクリーニングします。Ebstein病に合併するものはB型WPW症候群が多い。頻拍発作がなく,器質的心疾患や心機能低下がない場合は、運動制限なしで1~2年ごとの経過観察とする。頻拍発作についてはいたずらに不安がらせるのもよくないです。必ずしもみんながみんな頻拍を起こすわけではあ りません。ただし、頻拍を起こす可能性は普通の人よりは多いということで、 学校の先生とか家族は、検脈ができる、 脈がとれるようになるというのがまず第一だと思います。頻拍発作がある場合は、心房細動を合併すると小児でも稀であるが突然死のリスクがあり精査が必要である。(姫路市では、初回発作で突然死した症例があり、たつの市でもE可でフォローしている)
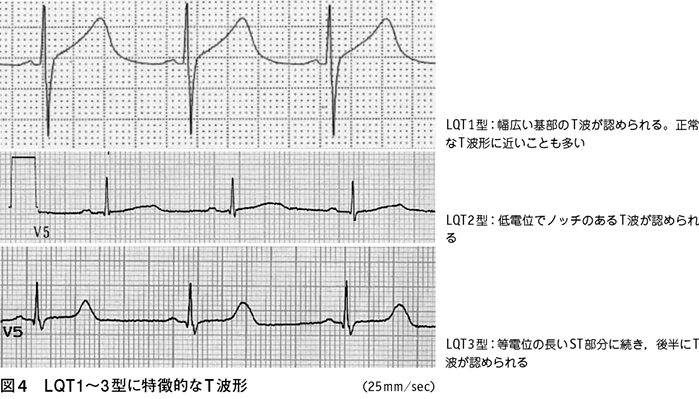
先天性QT延長症候群(LQTS)
遺伝性不整脈は,心臓の興奮・伝導を司る心筋イオンチャネルをコードする遺伝子に異常があり,心室頻拍・細動や致死的な徐脈を発症する家族性不整脈疾患である。代表的な先天性LQTSの頻度は一般人口の2500人に1人程度であり,学校検診で抽出すべき重要な疾患である。しかし20~40%の症例はQT時間が軽度延長にとどまり,変動もあるため,失神等の症状や家族歴の問診も重要である。2次検診以降への抽出基準のQTc値は、0.45秒以上とします。心拍数が速い小児においてBazett補正(QT/![]() )すると過剰補正されるため、Fridericia補正(QT/
)すると過剰補正されるため、Fridericia補正(QT/![]() )でQTc値を採用しています。2次検診以降では運動負荷心電図を行い、その結果をもとに運動制限を含めた管理指針を決定します。QTc≧0.50秒、失神、心停止または心室頻拍/torsade de pointesの既往、突然死の家族歴などがある場合は早期に専門医へ紹介します。
)でQTc値を採用しています。2次検診以降では運動負荷心電図を行い、その結果をもとに運動制限を含めた管理指針を決定します。QTc≧0.50秒、失神、心停止または心室頻拍/torsade de pointesの既往、突然死の家族歴などがある場合は早期に専門医へ紹介します。
遺伝子型は現在LQT1~15型まで知られているが,LQT1~3型で約90%を占める。それぞれ特徴的なT波形態を示します。発作誘因(リスク因子)にも特徴があり、LQT1型は運動(特に水泳)精神的緊張、LQT2型は目覚まし時計など突然の大きな音、精神的緊張、女性では出産後などが発作誘因となり、LQT3型では安静時、迷走神経緊張時の発作が多い。また,LQT1型,2型ではβ遮断薬が有効なことが多く、怠薬が大きなリスクになることを説明する。
先天性QT延長症候群(long QT syndrome:LQTS)(後述)も洞性徐脈傾向を示すことはよく知られている。
心筋症
2006年に米国心臓協会が提唱した心筋症の定義は「通常、不適切な心室の肥大や拡大を呈するような心筋の器質的あるいは電気的異常を有する多様な疾患群」であり、その原因は多岐にわたるが、しばしば遺伝性である。そして主な病変が心臓にあるものを原発性心筋症,全身疾患の心筋病変を二次性心筋症と大別している。欧州心臓病学会は,心筋症とは「心筋に構造的、機能的異常をきたす心筋障害であり、この障害を説明できるような冠動脈疾患、高血圧、弁膜疾患、先天性心疾患を有さないもの」と定義しており、形態・機能的異常をもとにした分類となっている。
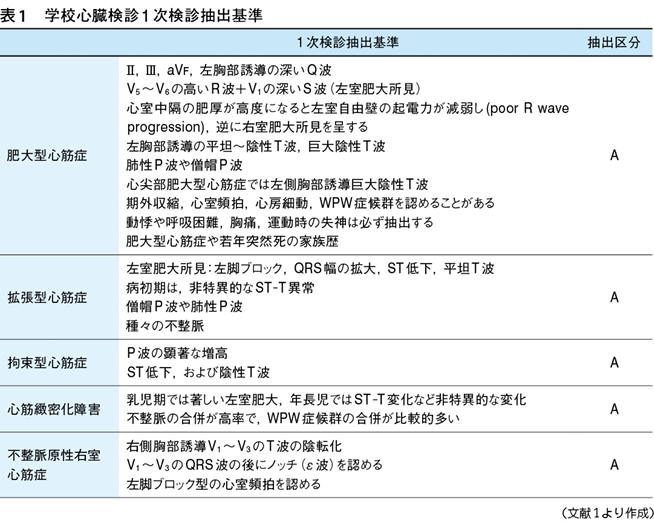
肥大型心筋症
著明な心筋肥大により心室内腔が狭小化し、拡張障害のため心室への流入血液量が減少し,心拍出量の低下をきたす。組織学的には錯綜配列を伴う心筋細胞の不均一な肥大を特徴とする。約半数に家族性が認められ,常染色体優性遺伝を示す。半数以上はサルコメアを構成する心筋収縮関連蛋白の遺伝子異常である。厚生省特定疾患特発性心筋症調査研究班が行った,病院へのアンケートによる全国疫学調査(1998年度)では、全国推計患者数は2万1900人、有病率は人口10万人当たり17.3人である。乳児期に1つのピークがあり,それ以降は10歳頃から発症するものが多い。肥大型心筋症は若年者の心臓突然死の最も重要な原因であり、特に運動中の突然死をきたすため、検診でのスクリーニングが重要である。小児期には無症状のことが多く,学校心臓検診が発見の契機となることがある。心室性不整脈による動悸,肺うっ血による呼吸困難,心筋虚血による胸痛を訴えることがある。Ⅲ音やⅣ音,左室流出路狭窄,僧帽弁閉鎖不全,右室流出路狭窄に伴う収縮期雑音を聴取することがある。
心電図上の特徴は以下の3点である。
①心室中隔の肥厚を反映してⅡ,Ⅲ,aVf、左胸部誘導の深いQ波がみられる。
②V5~V6の高いR波,V1の深いS波の左室肥大所見がみられる。心室中隔の肥厚が高度になると左室自由壁の起電力が小さくなり(poor R wave progression)逆に右室肥大所見を呈することもある。
③左胸部誘導の平坦〜陰性T波,巨大陰性T波,肺性P波や僧帽P波をしばしば認める。
ホルター心電図では肥大型心筋症の50~85%に心室期外収縮を,20~28%に非持続性心室頻拍を認める。30~50%に心房性頻脈性不整脈を認める。
心エコーでは左室心筋の肥厚が特徴的で,心室中隔の肥大(非対称性中隔肥厚,asymmetric septal hypertrophy:ASH)が著明なものが多い。左室流出路狭窄のあるものでは僧帽弁前尖の収縮期前方運動(systolic anterior motion:SAM)を認める。
肥大型心筋症では10歳頃から発症するものが多く、初発症状が運動中の失神や突然死のこともあります。若年突然死、心筋疾患の家族歴がないかよく聴取し、失神(特に運動中)、痙攣、胸痛、心停止あるいは持続性心室頻拍の既往歴がないか問診することが重要です。
突然死のほとんどは運動中であるため,運動制限を行います。無症状例では管理指導区分Dとする。胸痛や失神などの症状がある例および閉塞型の患児では,管理指導区分B/Cとする。


拡張型心筋症
心室の心筋細胞の変性や線維化の結果、心筋収縮不全により心拡大をきたし、心不全が進行する予後不良の疾患である。ウイルス性心筋炎との関連性が示唆されるものや,家族内発症における遺伝子変異例など,病因は多岐にわたる。18歳以下の小児を対象とした登録研究において10万人当たり1人/年となっている。小児期では、いずれの年齢にも認められ、5年生存率は50%と不良である。左心不全症状が主体で,乳児では哺乳不良,体重増加不良,顔色不良,末梢冷感が認められる。症状は、頸静脈の怒張,肝腫大,浮腫,心尖拍動の外側変位がみられる。胸部聴診では肺うっ血のため湿性ラ音や喘鳴を認める。房室弁逆流に伴う心雑音やⅢ音、Ⅳ音による奔馬調律を聴取する。
心電図上の特徴は以下の3点である。
①左室肥大所見:ST低下,平坦T波などがみられる。特に病初期は,非特異的なST-T異常であることも少なくない。
②僧帽P波や肺性P波を認める。
③種々の不整脈がみられやすい。
心房性、心室性不整脈は左心機能が低下するにつれて増加する。拡張型心筋症ではほぼ全例に心室期外収縮が,約半数に3連発以上の非持続性心室頻拍が認められ、多源性期外収縮も一般的に認められる。心エコーでの左室内腔の拡大と駆出率の低下は診断上有用である。進行とともに肥大の退縮、左室後壁基部の菲薄化、および限局性またはびまん性左室壁運動低下を示す。管理指導区分は、無症状ならD,有症状ならCとし、原則として競争的運動や学校の運動部は禁止する。
拘束型心筋症
心室の拡張や肥大はなく心筋の収縮力も正常であるが、一側または両側心内膜の高度の線維性肥厚、あるいは心内膜下心筋間質の高度の線維化により心室壁が硬化し、心室のコンプライアンスが低く、拡張不全をきたす疾患である。肥大型心筋症や拡張型心筋症に比べて稀であり,小児期心筋症の4.5%と報告されている。発症年齢も平均6歳と高く、他の心筋症に比べ乳児例は15%と少ない。予後は著しく不良であり、発症2年で50%が死亡する。強皮症、アミロイドーシス、サルコイドーシス等の全身性疾患に伴うもの、ムコ多糖症、好酸球増多症、悪性腫瘍や放射線治療に伴い発症するものがある。
呼吸困難や浮腫,肝腫大など,静脈圧上昇および肺うっ血に伴う症状が出現する。左房圧の上昇に伴う肺血管疾患は急速に進行することがある。心房細動をきたしやすい。肺高血圧をきたすとⅡ音の肺動脈成分が増強する。管理指導区分は、無症状ならD,有症状ならCとし、原則として競争的運動や学校の運動部は禁止する。
心筋緻密化障害
心室壁の過剰な網目状の肉柱形成と深い間隙を形態的特徴とし、心筋緻密層が低形成となり、心内膜下への血流供給低下による心筋虚血と菲薄な心筋緻密層による心収縮力の低下が惹起される遺伝的要素の強い心筋症である。X連鎖性のものやミトコンドリア遺伝子異常が疑われる家系があり、遺伝的多様性がうかがえる。新生児期,乳児期に重篤な心不全症状で発症する典型例から、徐々に拡張型心筋症様の症状を呈するもの、学童期に心臓検診で発見されるものまで症状は多彩である。いずれの年齢においても不整脈の合併が高率であるが,小児期ではWPW症候群や上室頻拍,完全房室ブロック,洞不全など先天性の要素が強い不整脈が多く認められる。年長児や成人では心室性期外収縮、心室頻拍や心房細動など心筋障害や線維化による二次的な不整脈の発生が主体である。臨床症候は拡張型心筋症の病態を呈するもの、壁在血栓による塞栓症を伴うことがある。心エコー検査による心室壁の著明な肉柱形成と深く切れ込んだ間隙の特徴的な形態が診断に有用である。
無症状で心機能正常例ではE禁または可(観察期間1〜3年ごと)とする。心機能低下例では,拡張型心筋症と同様,管理指導区分は無症状ならD,有症状ならCとする。5年で約半数が死亡あるいは心移植になる。
不整脈原性右室心筋症
主として右室心筋,時に両心室にみられる進行性の変性,脂肪浸潤,線維化を特徴とし,右室の拡大や収縮不全,右室起源の心室性不整脈を呈する進行性の疾患である。しばしば家族性に発生する。発症年齢は早くても思春期後期以後で、20~40歳がほとんどである。若年者の突然死の原因として重要で、初発症状のこともある。動悸、易疲労などの症状を呈するが、無症状のこともある。運動誘発性の心室性不整脈、頻発する持続性の心室頻拍により失神や突然死をきたす。
心エコーで右室に瘤形成,肥厚した肉柱,突出bulging,全体の収縮低下などの特異的所見を認める。MRIでは、右室の瘤形成、収縮低下、脂肪浸潤、遅延性濃染を認める。心筋生検で著明な脂肪浸潤,線維化を認める。予後不良を予測する因子は、若年発症、競技スポーツ選手、突然死の家族歴、広範な右室病変あるいは左室への進展、失神の既往、心室頻拍などであり、軽い運動を除き運動は禁忌である(C区分に相当)
先天性心疾患
通常、先天性心疾患(生まれつき心臓の形と機能に異常のある)あるの子供が産まれる確率は、約100人に1人と言われています。胎児の心臓は妊娠3週から8週にかけてできあがります。胎児の心臓ははじめ、1本の管として現れます。この管が曲がったり、ねじれたりして、塊になり、やがて、この中に仕切りができて、心房や心室、大血管や4つの弁などが完成します。この発達の過程の途中で、風疹など感染、薬物の副作用、放射線障害などを受けると、左右の心房や心室の壁が十分にできずに孔が残ったりするのです。しかし、実際のところは、妊娠期間中にこれといった心当たりがなくても先天性心疾患の子供は、一定の確率で生まれます。母親は第一に「自分に責任があったのでは?」と悩むこともあるでしょう。しかし、大半は原因不明と言われているのです。
先天性心疾患は、大きく分けて(1)非チアノーゼ性心疾患と(2)チアノーゼ性心疾患があります。
(1)非チアノーゼ性心疾患は、心臓に穴があいていて大量の血液が心臓と肺の間を空回りするために心臓や肺に負担のかかり、赤ちゃんは呼吸が速く苦しそうになり、汗をたくさんかき、ミルクがあまり飲めず、体重が増えないなどの症状が出てきます。風邪を引くと呼吸状態がさらに悪くなることがあります。
(2)チアノーゼ性心疾患は、酸素の少ない赤黒い静脈の血液が心臓の穴を通して大動脈から全身に流れるために唇や手足が紫色になり、体重の増加は比較的よいのですが、泣いたり、息んだり、熱を出したりしたときに全身が紫色になり、危険な状態になることがあります。
では、代表的な先天性心疾患の説明をします。同じ病名であっても病気の程度と進行度合いはお子さんひとりひとり異なりますし、二つ以上の病気が重なっていることもしばしばあります。
「おぎゃ〜」と生まれて
胎児は、母親から胎盤を通じて酸素を受けとっているので、肺はすっかりつぶれた状態でほとんど血流はありません。胎盤は、胎児の臍動静脈(臍の緒)を介して胎児と繋がっています。胎盤は、子宮に張り付いたようになっており、そこで膜を介して胎児の血液と母親の血液が物質交換(酸素や栄養)をします。胎児循環の特徴として、3つのバイパスがあります。胎盤で酸素や栄養を受け取った血液(動脈血)は、臍静脈を通り胎児に戻っていきます。臍静脈を流れる血液の大部分は静脈管(アランチウス管)を介して下大静脈へと注ぎ込まれます。血液は、下大静脈から右房へと流れ込みますが、右房では上大静脈から戻ってきた静脈血と臍静脈からの動脈血が混合されますが、臍静脈から戻ってきた酸素を多く含む血液(動脈血)のほとんどは、下大静脈弁の働きで右房と左房の間にある卵円孔という孔を通って左房に入ります。(一部は右房から右心室を通って肺動脈へ流れていきます)左房から左室に入った血液は、大動脈へ打ち出されます。酸素を多く含んだ血液は、大動脈から脳や上肢へ打ち出されていきます。一方で肺動脈に入った一部の血液(酸素の量が少ない)は、肺動脈と大動脈を繋ぐ動脈管(ボタロー管)を通り、下行大動脈へ流れ込んで、全身へ流れて行きます。そして内腸骨動脈から分岐した2本の臍動脈を経て胎盤へ戻ってきます。これが胎児循環です。胎児の脳や上半身へ行く血液は、たくさんの酸素を含んでおり、胎児の成長をみても脳や上半身の成長に比べ、下半身は遅れて生まれてくるのです。
赤ちゃんは、生まれると「おぎゃ〜」と産声をあげます。これは、肺に十分に空気が入り、その空気が吐き出された時の第一声です。これを合図に臍動静脈は無用のものになり、循環動態も大きく変化します。胎児循環で必要であった3本のバイパス(静脈管、卵円孔、動脈管)は機能的に閉鎖し、上大静脈、下大静脈から右房へ戻ってきた血液(静脈血)は、すべて右心室から肺動脈へ流れ、十分に膨らんだ肺で酸素をもらった血液(動脈血)は肺静脈から左房へ戻ってきて、左室から大動脈(全身)へ打ち出されていきます。

心室中隔欠損症

先天性心疾患の中で最も多く、約6割を占めるとされています。左室と右室の間を隔てている筋肉の壁である心室中隔に孔が開いている心臓病です。径が2〜3mmの孔が小さいものでは、2歳ぐらいまでに自然閉鎖が期待出来るので、経過をみます。圧格差が100mmHg近くになるので、生下時よりザーザーという大きな収縮期雑音(孔が小さいほど)が聴取されます。孔が小さくなる傾向は一生続くとされていますが、自然閉鎖しない場合も心臓への負担はほとんどありません。しかし、噴出するシャントの勢いは強く、菌血症を伴うような場合は、右室のシャントがぶつかる部位には、病巣(感染性心内膜炎)ができやすくなるので、抜歯などの際には予防的処置が大切です。5〜10mmの中程度の大きさの孔であれば、自然閉鎖は期待出来ず、心臓や肺に負担がかかり、乳児では呼吸が速くなり、ミルクの飲みが少なくなり体重の増え方も不良になり、一般的に手術となります。また、孔の場所によっては、大動脈弁の変形や、閉鎖不全をおこすこともあり、血液の漏れの量にかかわらず、手術になることもあります。手術は人工心肺を用いておこない、「パッチ」と呼ばれる人工のあて布を左右の心室の間にあてて、孔をふさぐという手術を行います。(孔をふさいだ後は、まわりの筋肉が成長するので患者さんが成長してもパッチの取替えをする必要はありません)大きな孔が開いている場合には、肺への負担が大きくなり、「肺高血圧」になります。左室と右室の圧は等しくなって、アイゼンメンゲル症候群(Eisenmenger)と呼ばれる状態になると手術ができないようになることもあります。

孔の開いている場所により4つのタイプに分類されています(Kirklin分類)
(1)漏斗部欠損(Ⅰ型): 右室流出路の肺動脈弁直下の欠損で、大動脈弁閉鎖不全症を合併しやすい。東洋系人種に比較的多い。自然閉鎖は稀です。
(2)膜性部欠損(Ⅱ型): 心室中隔の膜性部およびその周辺の欠損で、頻度は最も多い。自然閉鎖の傾向が強い。
(3)流入部欠損(Ⅲ型): 頻度は最も少ない。Down症に合併する頻度の高い心奇形のひとつである。
(4)筋性部欠損(Ⅳ型): 多孔性のことがある。頻度は少ない。自然閉鎖例が多い。西洋人に多い。
抗菌薬の予防投与
心疾患を有する患者さんに抜歯など口腔内処置を行う場合には、処置後の菌血症感染性心内膜炎を予防する目的で抗生物質を投与することがあります。AHAのガイドラインでは、抗菌薬の予防投与が強く求められている心疾患としては、人工弁置換術の患者、細菌性心内膜炎既往患者、複雑性心奇形(単心室、大血管転位、ファロー四徴症、Blalock-Taussig手術後)が挙げられ、予防投与が望ましい心疾患として、心室中隔欠損症、動脈管開存症、一次孔型心房中隔欠損症、各種弁膜症、肥大型心筋症などがある。(二次孔型心房中隔欠損症、心房中隔欠損症、心室中隔欠損症の術後6ヶ月以上経過、冠動脈バイパス術などは、予防投与は不要)起因菌として最も頻度の高いStreptococcus viridansが対象となり、抗菌薬としては、抜歯前日よりAMPC(サワシリン)1.5g/日、3〜7日間、ペニシリンアレルギーのある場合には、ファロム1.5g/日、クラリシッド400mg/日を代用している。
心房中隔欠損症

学校検診で発見されることがある代表的な疾患としては心房中隔欠損症がある。診断されない場合、主として成人期に心不全、不整脈、肺高血圧症を呈するようになるが、乳幼児期にこうした症状を呈するのは稀である。また、肺血流量の増加に伴い、胸骨左縁第2肋間に収縮期雑音を認める場合があるが、全例がこうした心雑音を呈するわけではないため、学童期まで未診断の症例が比較的みられる。
左房と右房の間を隔てている心房中隔という壁に孔が開いている心臓病です。全先天性心疾患の約1割を占め、心室中隔欠損症に次いで多い先天性心疾患です。
心雑音胸骨左縁第2~3肋間に収縮期駆出性雑音(相対的肺動脈狭窄)拡張期中期ランブル(相対的三尖弁狭窄音)Ⅱ音の固定性分裂(呼吸による変動がない)または、学校検診の心電図異常(不完全右脚ブロックパターン(IRBBB))などで発見される。
昔は、自然に孔が閉じることはないと言われていましたが、最近では、心エコーでわずかな孔まで診断されるようになり、約半数の症例で自然閉鎖すると言われています。8mm未満の孔なら1歳半までに自然閉鎖、あるいは穴が小さくなることも多いので、通常はしばらく経過観察しています。(1歳以上で10mm以上の孔であれば、自然閉鎖は難しい)しばらく経過観察しても3歳以降、大きな穴が開いている場合には手術が選択されます。肺高血圧がなく、肺/体血流比≧2.0なら、小児期から思春期の間に手術を行う。乳児期に心不全症状を起こしたものは、その時期に手術を行います。手術は人工心肺を用いて、心房中隔欠損の穴を閉じます。通常、子供の場合には直接縫い閉じることができることが多いですが「パッチ」と呼ばれる人工のあて布がつかわれることもあります。2006年よりカテーテル心房中隔閉鎖術が保険適用となり、経食道エコーによる評価が適切に行われれば、外科手術と同等の成績をおさめています。
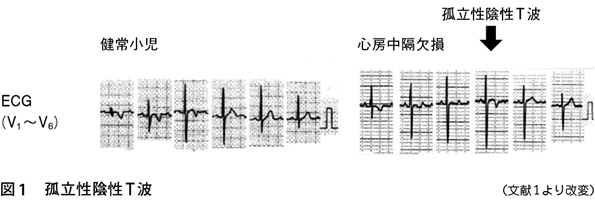
学校心臓検診の心電図所見としては,不完全右脚ブロック,V4誘導でのT波の陰転などで精密検査となることが多い。特に小児の胸部誘導においては,右側は陰性T波を呈することが正常で,年齢が上になるにしたがって,左側胸部誘導から順に陽転化していく。しかしながら,心房中隔欠損症においては,図1のように,V4誘導でのT波は,それよりも右側のV3誘導が陽性T波であるにもかかわらず,陰性化している特徴的な「孤立性陰性T波」を呈する場合があるので、こうした症例は不完全右脚ブロックを呈していなくても,心エコー検査で心房中隔欠損症の有無を検索が必要である。他の所見として右軸偏位やⅠ度房室ブロックを伴うこともある。
放置した場合の臨床症状と経過は、乳幼児期は無症状で、20代後半より労作性呼吸困難、動悸、息切れで発症します。合併症の肺高血圧の多くは思春期以降に生ずる。僧房弁逸脱症は、15~40%に出現。感染性心内膜炎の合併はきわめて稀である。

【分類】
① 二次孔中隔型 secundum type: 最も多い。(男女比 1:2)
② 一次中隔型 primum type
③ 静脈洞型 sinus venosus type
④ 冠静脈洞型 coronary sinus type
◎部分肺静脈還流異常症: 9%に合併し多くは静脈洞型に認められる。
肺動脈狭窄
肺動脈狭窄には、肺動脈弁下狭窄、弁狭窄、弁上狭窄、末梢狭窄があり、最も多いのは肺動脈弁狭窄です。フォロー四徴症や大血管転位症などに合併しておこりますが、この疾患のみが単独でくることもあります。肺動脈の狭くなっている部分で血流が早くなり、収縮期の心雑音で発見されます。心エコー検査で、その部分の圧較差を測ることで重症度がわかります。あまりにも狭い場合は、右室に負担がかかるので治療が必要になります。最近は、圧較差が40mmHg以上でバルーン肺動脈弁形成術(PTPV)の適応になります。
動脈管開存
動脈管は、胎児循環では必要不可欠なものですが、生後、次第に肺呼吸を開始したことによって、肺が柔らかくなって肺の血管抵抗が下がり始めると、普通は、生まれて数日以内に閉じてしまいます。しかし、動脈管が生まれた後も閉じないで、残ってしまった場合(動脈管開存と言います。未熟児に多く発生します。)血液は大動脈から肺動脈へ流れ込むようになり、左右の肺へ流れる血液量、さらに左房、左室に泣かれる血液量が増えることで心不全を起こし、呼吸が苦しくなったり、ミルクが飲めなくなったりします。連続性の心雑音や心エコーで診断されます。治療としては、インドメタシンで動脈管を閉じることができる場合もありますが、手術による閉鎖術(結紮術または離断術)が行われます。最近では、胸腔鏡によるクリッピングやカテーテルを使ったコイル塞栓術なども行われています。
心内膜床欠損症 (房室中隔欠損症)
房室中隔欠損症は、従来、発生学的な見地から心内膜床欠損症と呼ばれていたものが、最近は解剖学的見地から房室中隔欠損症と呼ばれるようになったものである。この疾患は、完全型と不完全型があり、両者の違いは前尖と後尖がconnecting tongueで結合していて,僧帽弁口と三尖弁口が分離しているものが不完全型、前尖と後尖がconnecting tongueで結合しておらず、僧帽弁口と三尖弁口が共通房室弁口になっているものが完全型である。一般的に不完全型は心房中隔の一次孔のみが欠損しており,完全型は一次孔欠損に心室中隔欠損を伴うことがほとんどである。房室中隔欠損の約半数は、ダウン症候群に合併しています。心内膜床から形成される部分には、心房中隔、心室中隔と三尖弁、僧帽弁が含まれているので、心房、心室に孔が開くだけでなく、三尖弁閉鎖不全や僧帽弁閉鎖不全も合併します。これらの形成不全の程度によって、完全型から不完全型などに分けられていますが、完全型では、生後1ヶ月ぐらいから肺へ流れ込む血流量が増加して、肺高血圧を合併して早期にアイゼンメンゲル化するため、心内修復術を行います。
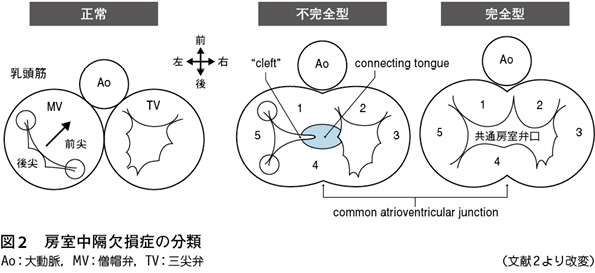
房室中隔欠損症は,心房間,心室間それぞれの欠損孔におけるシャント量や,僧帽弁,三尖弁の逆流量などにより程度が異なるが,聴診上,胸骨左縁上部の収縮期雑音,または胸骨左縁下部の全収縮期雑音や拡張期ランブル,心尖部全収縮期雑音や拡張期ランブルを認める。肺高血圧を伴う場合はⅡ音の亢進を認める。心電図所見としては,左軸偏位が特徴的であり,不完全右脚ブロックを認めることが多い。房室中隔欠損症は,通常は学校心臓検診を受ける年齢までには診断されているが、不完全型房室中隔欠損症は稀に学校心臓検診で発見される場合がある。
また,不完全型房室中隔欠損症と心房中隔欠損症の鑑別は、心房中隔欠損症では多くは二次孔欠損型であるのに対し、不完全型房室中隔欠損症は、一次孔欠損で欠損孔の位置が異なることのほか、僧帽弁と三尖弁の心室中隔への付着位置が正常あるいは心房中隔欠損症とは異なり、房室中隔欠損症では心尖部から見て同じ高さにあること、房室弁形態の異常の有無などから判断できる。なお、房室中隔欠損症は21トリソミーにおいて、比較的頻度の高い先天性心疾患であることも知っておくとよい。
大動脈縮窄
大動脈の一部に狭い部分ができた心臓病です。大動脈は、心臓から出た後、首の下で弓形に反転して腹部から下半身に向かって走っていきます。狭窄部は、大動脈弓で、腕頭動脈、左総頸動脈、左鎖骨下動脈の3本の血管を分枝した直後から動脈管の接続部までの範囲に生じます。狭さの度合い(重症度)によって症状の出る時期が異なります。重症例(圧較差が大きい)では、生後2〜3ヶ月で血圧の上昇による心不全を起こしてきます。軽症例では学童期や青年期で気づかれることもあります。(手と足で血圧の差が生じる)治療は、カテーテル治療と手術があります。
大動脈縮窄複合・大動脈弓離断複合
大動脈縮窄症は大動脈弓遠位部の狭窄を呈する疾患で,心室中隔欠損や大血管転位、動脈管開存などを伴う複合型と,それらを伴わない単純型がある。同じ組み合わせで、大動脈縮窄の部分が完全に離れてしまっている場合は、大動脈弓離断複合と言います。複合型は多くは、生まれた直後は一見元気なのですが、顔は赤いのに、足は紫色(チアノーゼ)になり、泣くといっそう顕著になります。この病態で、生後数日目に動脈管が閉じてしまうとショック状態に陥います。すぐにこの病気と診断がつけば、プロスタグランジンを点滴して、動脈管を再開通することでショック状態から回復させることができます。動脈管開存で、肺血流量が増えて、ミルクが飲めない、呼吸が苦しいなどの症状が発症する前に、それぞれの病態に対する手術を行わなければなりません。早期に手術を要する疾患であるため,学校検診で初めて診断されるということはないが,単純型は成人期まで自覚症状がない場合もあり,学校心臓検診で見つかる可能性がある。単純型大動脈縮窄症では,血管雑音が聴取される場合があるほか,大動脈弓の狭窄部より近位部に通常は腕頭動脈,左鎖骨下動脈があるため,上半身が高血圧となり,狭窄の程度によっては,心電図上も左室の圧負荷所見がみられることがある。
この疾患は脳出血,冠動脈硬化,心筋梗塞などの合併につながりやすく,特に学校心臓検診で高血圧を指摘された場合は,鑑別疾患として挙げる必要があることを認識しておくべきである。なお,大動脈縮窄症では大動脈弓からの分枝異常を合併する場合がある。たとえば,縮窄近位部から起始する左鎖骨下動脈より腕頭動脈が遠位,かつ縮窄部より遠位から起始している場合,左上肢で測定すると高血圧であっても,右上肢は血圧が高くないということが起こりうる。そのため,若年の高血圧では両上肢と下肢で血圧測定をすることが望ましい。
大動脈肺動脈中隔欠損
心臓が発生する過程では、肺動脈と大動脈はもともと1本の大きな血管ですが、真ん中に螺旋型の壁ができて2本に分けられます。この壁の形成がうまくいかず、壁の孔が残存したままになるとこの病気になります。生後1ヶ月ぐらいから肺動脈へ流れ込む血液が増えて、ミルクが飲めない、呼吸が苦しいなどの症状が出てきます。この疾患には、高頻度に大動脈縮窄などの異常を伴いますので、他の奇形の合併がないか注意が必要です。
大動脈弁狭窄
高齢者にはなじみの弁膜症ですが、大動脈弁が生まれつき狭い疾患です。重症度によって緊急性が違います。
(1)新生児重症大動脈弁狭窄
生まれた直後から元気がなく、足にチアノーゼを認め、左室の収縮力はほとんどなく、放置すると尿がでなくなり、数日で死亡する重症の心疾患です。最近は、胎児エコーで診断し、帝王切開後なるべく早期にバルーンカテーテルで弁を拡げる治療を行います。数回繰り返し行う場合もあり、結果、重症の大動脈弁逆流を起こしてしまった場合は、Ross手術の適応になります。
(2)大動脈弁狭窄
左室に高い圧がかかり、左室肥大が起こってきます。成人と同じで左室と大動脈の圧較差が50mmHg以上になれば、治療の適応となります。バルーンカテーテルによる方法と手術(大動脈弁形成術、人工弁置換術、Ross手術など)があります。
ファロー四徴症
Fallotはフランスの医師の名前です。手足や口唇が紫色になるチアノーゼを生じる代表的な先天性心疾患です。ほとんどの患者さんは、生後1ヶ月以内に心雑音やチアノーゼで発見されます。乳幼児が、寒い冬の朝方、哺乳後、強く泣いた後、入浴後などにチアノーゼが強くなって呼吸困難が出現し、時には意識消失やけいれん発作を起こします。(無酸素(低酸素)発作)発作が起こりかけたら赤ちゃんの膝を曲げて、胸につけるように折り曲げた体制で抱いてあげると効果的です。無酸素発作を起こすのは、右室から肺動脈への通路が過収縮するためです。薬のよって発作が予防できなかったり、チアノーゼが強い場合は、手術が必要です。我が国では、1歳を過ぎてから根治術を行う施設が多いようですが、1歳までは、通常、鎖骨下動脈と肺動脈をバイパスで繋ぐ手術(Blalock-Taussigシャント手術(BTシャント))を行います。
完全大血管転位症
右心室から大動脈が、左心室から肺動脈が繋がる心臓病です。酸素含有量が低い静脈血が右心房から右心室を経由してそのまま大動脈へ出て全身へ循環するため、ほとんどは生後ただちにチアノーゼがあることに気づかれます。胎児循環で必要な心房中隔にある卵円孔が開存していれば、左房にある動脈血が右房に入り静脈血に混ざって全身に循環するために生存することができますが、卵円孔が生後すぐに閉じてしまった時は、直ちにバルーンカテーテルで心房中隔に孔をあける心房中隔裂開術(BASバス)が行われます。根治的な手術は、合併する他の心疾患によって異なりますが、肺動脈狭窄を合併していない場合は、肺動脈と大動脈を入れ替えて縫い付け、冠動脈を移植するジャティーン手術が行われることが多いようです。
修正大血管転位症
右心室から大動脈が、左心室から肺動脈が繋がる心臓病なので、大血管転位症と同じなのですが、右心室と左心室の位置が正常と入れ替わっているので、血液の流れとしては、静脈血は右心房→左心室→肺動脈へ、肺で酸素化された動脈血は、左心房→右心室→大動脈へと流れていきます。心室中隔欠損や肺動脈狭窄を合併していない場合は、血液が流れだけをみると正常と同じで(修正という意味です)チアノーゼも認めず、心雑音もなく、発見されずに成人になっている人もいます。心臓は胸郭の真ん中から右にあることが多く、心室中隔欠損と肺動脈狭窄を合併した場合にはチアノーゼが出現し、その他、三尖弁閉鎖不全、完全房室ブロック、WPW症候群に伴う発作性頻拍症などを合併して、徐脈や心不全を起こしてきて、ペースメーカーの植え込み術や心臓の手術が必要になってくる症例もあります。
両大血管右室起始症
正常では、肺動脈は右心室から、大動脈は左心室から起始しますが、その両方の血管ともが右心室から起始する疾患です。
いずれのタイプであっても全身に血液を送り出す大動脈が右心室から出ているわけですから静脈血が全身に駆出されることによるチアノーゼが出現します。肺動脈狭窄の有無も
肺動脈狭窄がなくて肺高血圧を伴うタイプでは、高頻度に大動脈縮窄を合併します。このようにいくつもの奇形が組み合わさっていることが多く、病態に基づいて、正確な診断ジャティーン手術やラステリー手術など適切な
総肺静脈灌流異常症
左右の肺で酸素化された血液は4本の肺静脈ですべて左房に戻ってくるのが正常ですが、この4本の肺静脈すべてが、左房へ繋がらないで、種々のルートを経由して右房へ繋がる心臓病です。ルートに毛細血管が入っていたりすると強い抵抗により、生後1ヶ月以内に、急激に呼吸困難になり、強いチアノーゼを呈し、手術が間に合わなければ、そのまま死亡してしまいます。
三尖弁閉鎖症
右心房から右心室への通り道である三尖弁が生まれつき閉鎖している疾患です。血液の流れ込みがないため、右心室は非常に小さくなり、ポンプとしての働きをすることはできないため、ポンプは左心室1個だけなので、単心室の仲間の心臓病です。このような心臓の小児からチアノーゼをなくす方法として、1970年、フランスの医師Fontanが、フォンタン手術を考案しました。上半身と下半身から戻ってきた静脈を直接肺動脈へ繋いで、心室から肺動脈への繋がりは遮断してしまう手術です。ただし、フォンタン手術が成立するためには、心室のポンプを使わないで、肺動脈に静脈血を流さなくてはなりません。そのため、肺高血圧を合併しているような赤ちゃんでは、肺動脈の壁が厚くなってしまう前に肺動脈にバンドをかけて、肺動脈の圧を下げるバンディング手術(肺動脈絞やく術)を受けておく必要があります。
エプスタイン奇形
右房と右室の間にある三尖弁が生まれつき、僧帽弁に比べ、極端に低い位置に付着するため、三尖弁が閉鎖せず、重症の三尖弁閉鎖不全を合併する心臓病です。エプスタイン奇形の重症度は、ピンキリです。最重症は、生まれてくるまでに心不全で亡くなってしまいますし、無症状で天寿を全うする人もいます。エプスタイン奇形の30%の人にはWPW症候群を合併します。心房中隔欠損症を合併していることもあります。
左室低形成症候群
10000人に1人の割合で生まれる稀な先天性心疾患で、以前はほとんど助からない心臓病としてほぼ諦められていましたが、最近は救命できるあかちゃんも増えてきました。この疾患は、生まれた直後は、比較的チアノーゼも目立たず、元気なあかちゃんのように見えるのですが、数日から1週間以内に状態が急変し、ショック状態になってしまうのが、この病気の特徴です。左心室が小さく、大動脈も2mmぐらいと細くなっています。右心室と肺動脈は立派で、生まれた後も全身には右心室から肺動脈、動脈管をとおして、全身(上半身にも細い大動脈を逆行して)に血液を供給しています。つまり、胎児循環がそのまま継続されているわけですが、動脈管が閉じてしまうととたんに血圧が下がって、ショック状態になるわす。手術で積極的に治療する場合は、3回に分けて手術を行います。まず、最初の手術は生後1週間〜1ヶ月までの間に、肺動脈を左右の肺動脈に分岐する根元で切り離し、右心室に繋がっている肺動脈と肺動脈弁を使って細い大動脈弓を太く形成します。切り離した肺動脈主幹部と右心室を細い人工血管でバイパスします。(ノーウッド手術)第2期の手術は、生後3ヶ月ころから右心室からの細い人工血管を閉鎖して、上大静脈を右肺動脈にバイパスする手術です。(両方向性グレン手術)第3期手術は、2歳半〜3歳ごろに行います。下大静脈も肺動脈に吻合し、フォンタン手術を完成させます。これで、すべての静脈血が直接肺動脈に流れ、チアノーゼは消失します。
単心室
単心房
無脾症候群・多脾症候群・内臓心房錯位症候群
ここには、代表的な先天性心疾患だけを取り上げましたが、実際にはいろいろな組み合わせの奇形が100種類以上存在し、それぞれの子供たちの血行動態を考えながら治療に当たらなければなりません。先天性心疾患は、生まれてくるあかちゃんの1%(100人に1人)の確率で起こるといると言われているので、年間100万にのあかちゃんが生まれるとすると1万人の先天性心疾患のあかちゃんがいるということです。最近の小児の心臓手術の技術の進歩は目覚ましく、一部のどうしようもない患者さん以外は救命できるようになったので、2000年には、小児(15歳以下)と成人の先天性心疾患の数が同じになったようで、今後は、成人の先天性心疾患の方が多くなり、われわれ内科医が小児科医にかわって先天性心疾患の術後を診ていかなければならない時代になっているわけです。
ASDは、成人の先天性心疾患の3割を占める。40歳までに手術をした方がいい
その他
複雑な先天性心疾患の多くは,術後例も含めて,専門機関で管理されることが多いが,管理区分に重要な影響を与える遺残病変の有無,肺高血圧症の有無,不整脈の有無などを適切に評価した上で,適切な運動管理区分を決定する必要がある。1つひとつの疾患の管理区分の決定は本項で解説するだけの紙面がないため,日本循環器学会/日本小児循環器学会合同ガイドラインの「学校心臓検診のガイドライン」(2016年版)3)などを参照して頂きたい。
4. 学校心臓検診に関連するガイドライン
学校心臓検診において,特に先天性心疾患について参考にすべきガイドラインを最後にまとめる。まず,日本循環器学会/日本小児循環器学会合同ガイドラインの「学校心臓検診のガイドライン」(2016年版)3)は,1次検診から診断,管理に至るまで網羅されたものである。また,1次検診の心電図検診に携わる場合,必須なのが日本小児循環器学会からの「学校心臓検診 二次検診対象者抽出のガイドライン」(2006年改訂)4)である。日常診療の診断基準と,検診でのスクリーニングとで,精査とする基準は異なるため,精査の対象者をできるだけ適切に抽出するために,ぜひ目を通して頂ければと思う。
診断後の管理方針については日本循環器学会の「心疾患患者の学校,職域,スポーツにおける運動許容条件に関するガイドライン」(2008年改訂版)5),「先天性心疾患術後遠隔期の管理・侵襲的治療に関するガイドライン」(2012年改訂版)6),日本小児循環器学会の「先天性心疾患の学校生活管理指導指針ガイドライン」(2012年改訂版)7)が参考になる。これらのガイドラインはすべて日本循環器学会あるいは日本小児循環器学会のウェブサイトより,非会員でも無料でダウンロードできるので,ぜひ役立てて頂きたい。